Grounded in 250M+ peer-reviewed papersSee how →
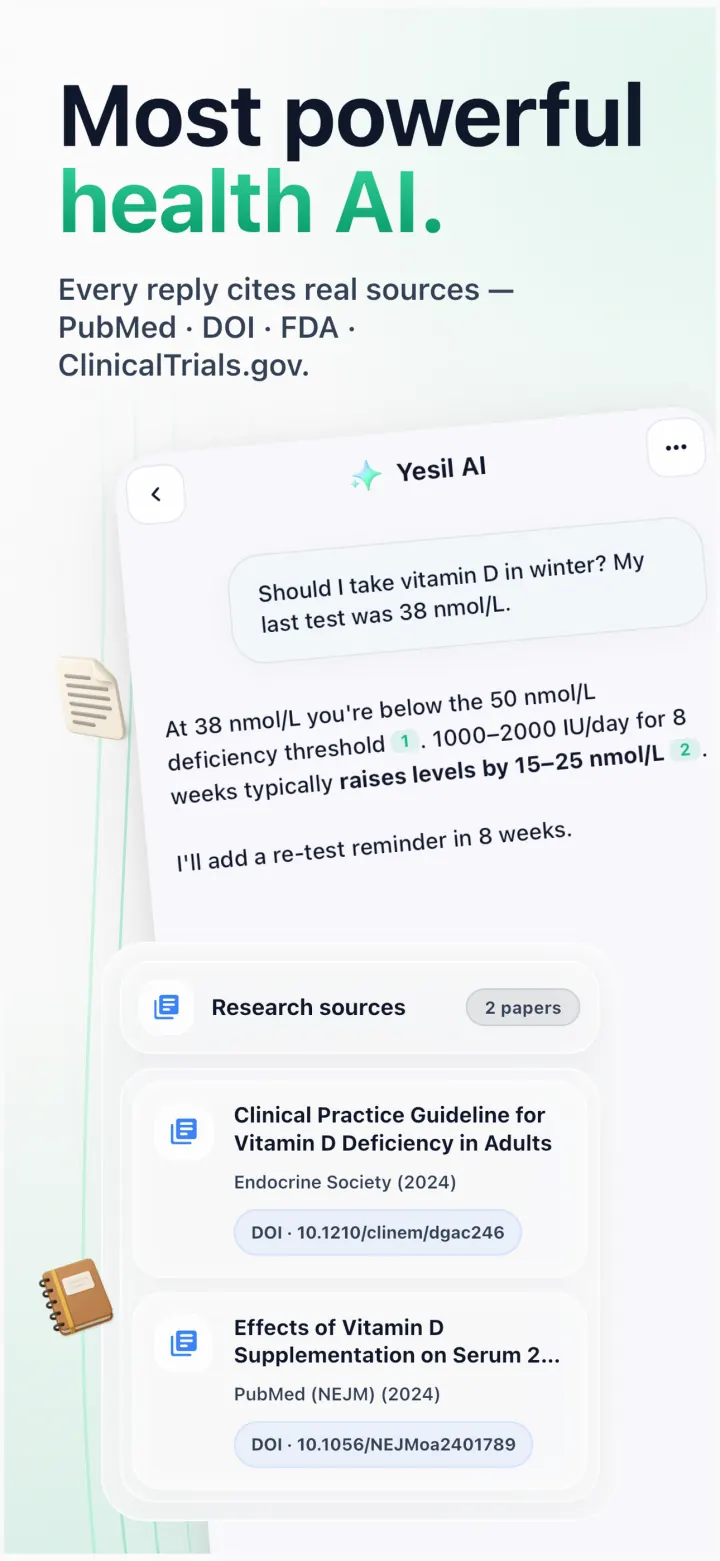
Reads your 360° health data. Cites real evidence. Remembers what matters.
 Cited evidence
Cited evidenceNEJM · 2024
DOI 10.1056/NEJMoa2401789
 Steps8,247
Steps8,247 Sleep7.4 h
Sleep7.4 h BP120/80
BP120/80 Remembered
Remembered“Your ferritin was 18 ng/mL — time to re-test.”
from 3 months ago
ReasoningGrounded in
Clinician? Get the professional version at
app.yesilhealth.comProSee it in action
One health AI that connects every program, every lab result, every symptom — and turns it into a daily plan you can actually follow.
 01
01Tell your AI coach what matters — sleep deeper, lose weight, lower stress. It builds a daily plan and adapts as you progress.
 02
0225 programs plus Apple Health and Google Health Connect in one dashboard. Log a meal in a single sentence — macros calculated in seconds.
 03
03Sees every program, every lab result and every saved memory — and adjusts your targets, challenges and daily plan. Not just answers.
Just talk. Ask a question and get a real answer — not a generic search result. Every important recommendation links to its source, and when the evidence is weak, the app says so.
250M+ research papers
Every indexed peer-reviewed article
Every FDA drug label
Approved monographs and safety data
All registered trials
Every NCT-registered clinical study
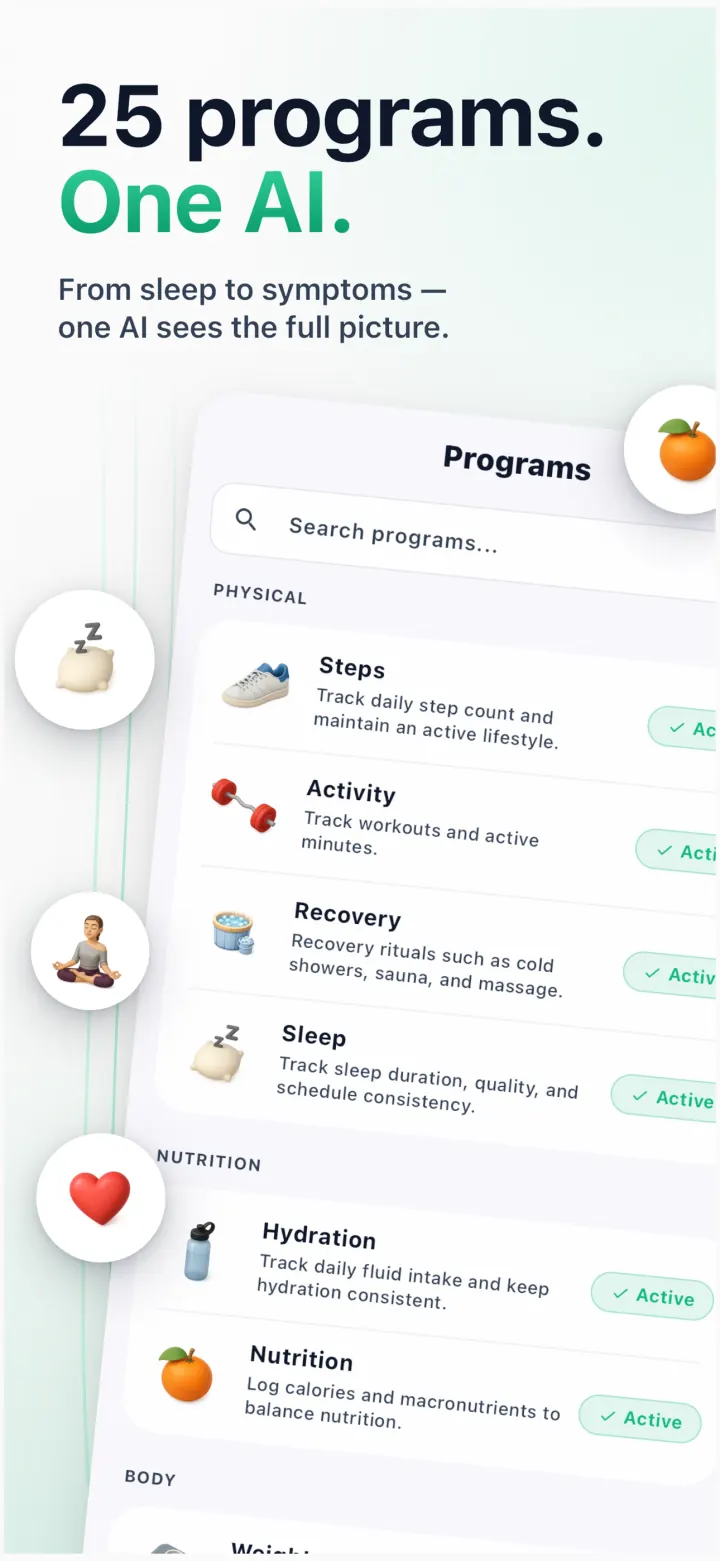
Every program shows a clean progress ring, a baseline-shift chart and AI insights you can act on. Pick what matters to you.
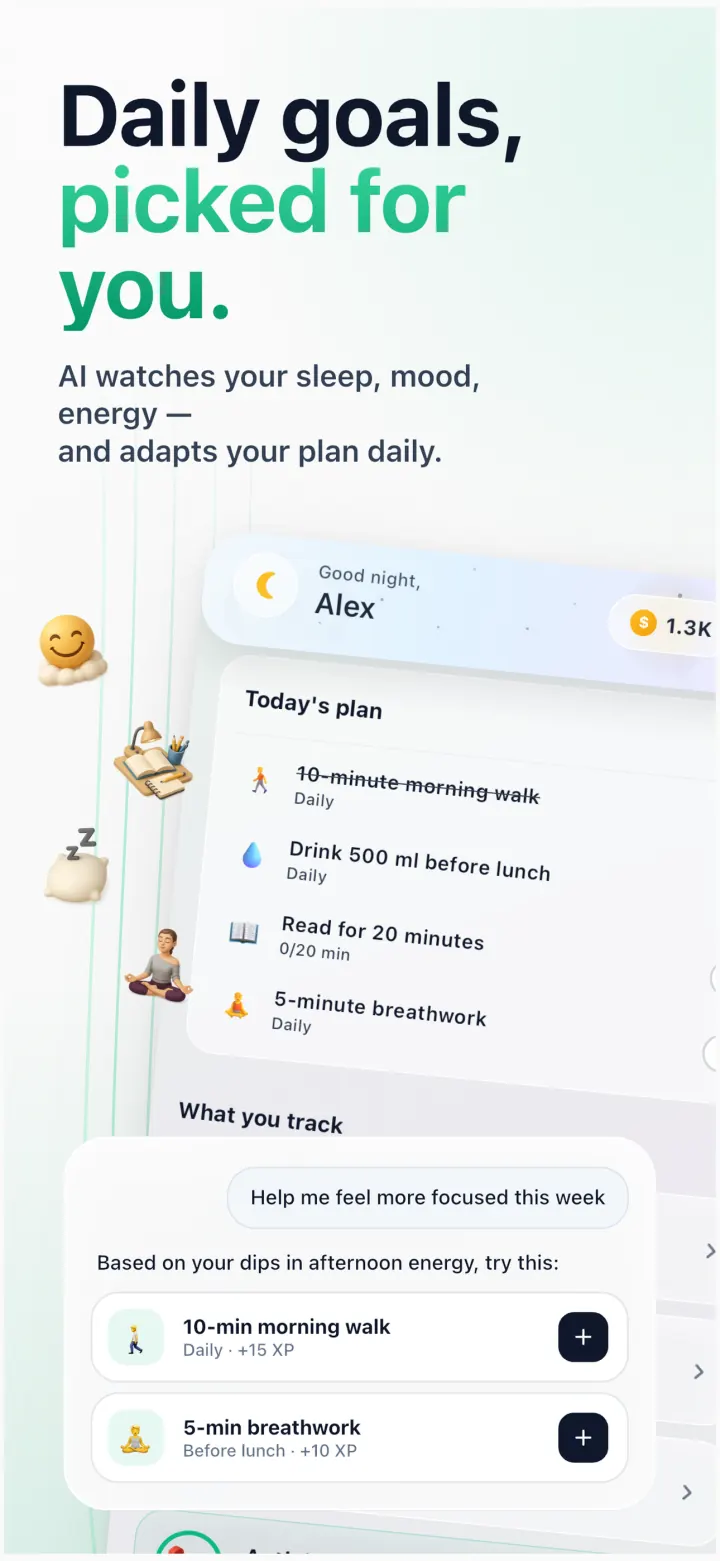
Science-backed micro-actions tied to your programs — “8-hour sleep window”, “no added sugar”, “evening walk”. The AI watches your patterns and adapts the plan daily.
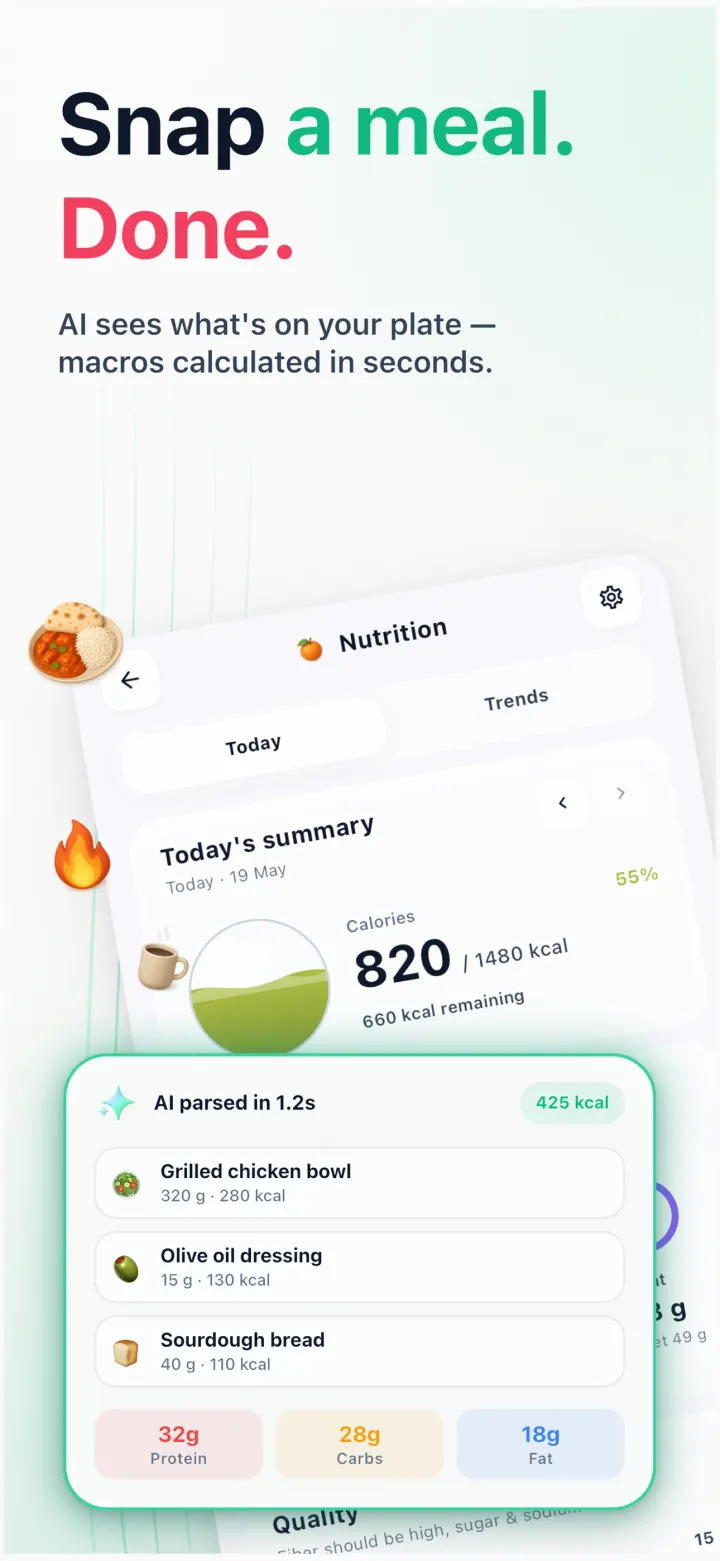
No tedious food databases. Take a photo or type one sentence — “chicken salad for lunch” — and the AI calculates calories and macros in seconds.
Paste your chat history from any AI. Yesil keeps only the health signals — and turns them into persistent memory.
ChatGPT
chat export
Claude
chat export
Gemini
chat export
Perplexity
chat export
Health-relevant signals only
Yesil Memory™Hypertension, diabetes, thyroid — long-term tracking, medication adherence and trend-aware nudges.
Tired all the time? See what your sleep, mood and recovery data actually say — and what to change first.
Recovery scores, HRV, sleep windows and load — synced from your wearable, interpreted in context.
Upload labs, see what's actually outside range, and get evidence-based next steps — with citations.
“It flagged my low ferritin three months before my GP would have. Every answer cites its source — that's what made me trust it.”
Maya
Runner · Singapore
“I walk into the visit already knowing what they've been tracking. Saves me 15 minutes and we focus on the right things.”
Dr. K.
Family physician · Berlin
“Dropped the API into our app in a weekend. Cited evidence on every response was the unlock for our compliance team.”
Daniel
CTO · health-tech startup · Boston
Trusted by clinicians, developers and consumers
One engine, three audiences. Same evidence — cited, calibrated and ingested daily.
 In early access with design partners
In early access with design partnersLicense Yesil Health Intelligence — the same evidence-grounded engine that powers the Yesil App and Yesil for Clinicians. 250M+ peer-reviewed papers, every FDA label, every registered trial. Cited on every response.
const stream = await yesil.chat.stream({question: "Should I take vitamin D this winter?",demographics: { age: 38, sex: "f" },health_context: "25(OH)D 18 ng/mL · 6.2 h sleep…",}); // SSE: token · citation · memory_extracted · donefor await (const e of stream) {if (e.type === "citation") {// { source: "NEJM", doi: "10.1056/…" }}}One web app for clinicians and developers — ask clinical questions against 250M+ peer-reviewed papers, or generate an API key to build on the same engine. Sign in at app.yesilhealth.com — your first 5 queries are free.
Yesil Web is live — clinicians and developers, one sign-in.
Informational support for clinical decisions. Not a medical device.

Health data is sensitive. We treat it that way — by default.
Your data is encrypted in transit and at rest, on managed infrastructure with role-based access.
We don't sell your data and don't train third-party foundation models on it without your permission.
Review and delete your memories, chats and account anytime. Sign in with Apple supported.
Keep tracking your programs for free. Unlock the full AI with a 3-day trial — no auto-charge surprises.
For staying on track
or $68.99/yr
5× more Health AI access
or $199/yr
No. Yesil gives you educational, evidence-based health information and helps you track and understand your habits. It does not diagnose, prescribe, or replace professional medical care. Always consult a qualified clinician for medical decisions.
Important recommendations cite their sources — peer-reviewed papers from the entire indexed medical literature (250M+ articles), FDA drug data, and every registered clinical trial. When the evidence is weak or mixed, the app tells you so instead of guessing.
Your health data is encrypted in transit and at rest. We don't sell it, and we don't train third-party foundation models on it without your permission. Sign in with Apple is supported.
Yesil Health runs on iOS and Android, syncs with Apple Health, Google Health Connect and Bluetooth devices, and speaks 9 languages: English, Türkçe, Español, Deutsch, Français, 日本語, 한국어, العربية and 中文.
Yes — sign in at app.yesilhealth.com for chat and the developer API, with the same account and memory as the app. The mobile app adds full tracking, programs and Apple Health.
Yes. Start with a free trial — no auto-charge surprises. You can cancel before it ends and keep using the free program tracking.
The same engine clinicians use, now in your pocket. Free to start.
